Janvier 2006 - n°106
Fractionnement par Couplage Flux Force associé à la diffusion de la lumière multi-angles : une solution viable pour un repliement protéique haut rendement
Auteurs : Christoph Johann, Wyatt Technology, Paul Ramage & René Hemmig, Unité Structure Protéique, Technologies centrales, Novartis Pharma AG
Introduction
Depuis l’achèvement du Projet Génome Humain, au cours
duquel environ 100 000 protéines humaines ont été identifiées,
un grand nombre d’entre elles ont été mises en évidence
comme des cibles potentielles par les sociétés pharmaceutiques
et biotechnologiques. Ces sociétés ont besoin de protéines
de haute qualité en tant qu’outils dans le cadre du développement
de tests, d’études structurelles des protéines et afin
d’identifier les fonctions biologiques.
Cependant, parmi ces protéines cibles, moins de 500 sont effectivement
disponibles dans le commerce. Le problème auquel les chercheurs sont
confrontés consiste à obtenir des quantités suffisantes
d’un grand nombre de protéines, à la fois rapidement et
à un coût raisonnable.
Ce besoin d’une production à grande échelle de protéines
recombinantes nécessite que les protéines se trouvent dans une
conformation native et active (c’est-à-dire repliée).
Comme les fonctions biologiques des gènes et des protéines correspondantes
sont inconnues, il est impossible de déterminer ci ces derniers se
trouvent dans un état natif ou non natif par l’intermédiaire
d’un essai biologique.
Les méthodes alternatives de contrôle du repliement protéique,
par chromatographie liquide haute performance en phase inverse (RP-HPLC),
chromatographie d’exclusion stérique (SEC) et fluorimétrie
présentent de nombreuses limites. Le présent article se propose
d’étudier l’utilisation du fractionnement par couplage
flux-force (Field-Flow Fractionation, FFF) associé à la diffusion
de la lumière multi-angles (FFFMALS) par l’Unité structure
protéique de Novartis Pharma AG, Bâle, Suisse, en tant que méthode
viable de caractérisation des protéines repliées.
Repliement protéique à haut rendement
L’Escherichia coli (E. Coli) demeure l’expression de choix pour
les études structurales. Cela est du à l’homogénéité
des protéines produites, à leurs niveaux d’expression,
à la rapidité de fabrication et à la facilité
de marquage. Cependant, pour un nombre donné de cibles, un certain
pourcentage sera soluble et le reste insoluble (entre 40-60 %), appelé
matériaux insolubles et connus comme des corps d’inclusion (IB).
Il est facile d’isoler, d’extraire et de purifier ces IB par le
biais d’un processus appelé la solubilisation ; le problème
toutefois réside dans l’extraction de protéines fonctionnelles,
correctement repliées à l’aide d’un repliement à
haut rendement.
Le repliement est un processus résultant en un changement de la conformation
de la protéine de l’état déplié à
replié (natif). Afin d’y parvenir, les IB sont initialement solubilisés
par des agents comme des détergents, des chaotropes ou de l’urée.
Les IB solubilisés contiennent des impuretés, lesquelles peuvent
interagir avec la protéine exprimée et ainsi interférer
avec son repliement. En outre, le fort degré de concentration du dénaturant
utilisé pour solubiliser les IB rend les protéines désordonnées
(dépliées), dissoutes et flexibles. Par conséquent, il
est nécessaire de réduire la concentration de dénaturant
et de modifier les conditions de la solution d’une manière contribuant
au processus de repliement. Le processus visant à trouver le meilleur
protocole est basé sur des essais et des expériences erronées,
lors desquels sont testés un ensemble de produits chimiques et de conditions.
Méthodes de lecture du repliement
Un certain nombre de variables peut être inclus afin de maximiser les
chances de succès du processus de repliement, par exemple des ajouts
de tampon, des pH différents et des chaperons artificiels. Cependant,
une fois que le repliement a eu lieu, la difficulté consiste à
déterminer s’il a été réussi, par exemple
en faisant la différence entre une solution optiquement transparente
contenant des protéines repliées et une solution transparente
contenant des protéines dénaturées ou agrégées.
Des techniques comme la RP-HPLC ou la turbidimétrie ont peu de chances
de distinguer les deux, la spectroscopie du dischroïsme circulaire (CD)
est insensible et fournit un faible rendement. La fluorimétrie, par
contre, peut différencier des protéines repliées et dépliées,
mais nécessite des concentrations protéiniques supérieures,
non compatibles avec un format de plaque à 96 puits. De plus, la sensibilité
dépend fortement du contenu de résidus tyrosines et tryptophanes
dans la protéine, ainsi que des effets d’atténuation causés
par certains des additifs tampons utilisés. Par conséquent,
la méthode de fluorimétrie ne fonctionne pas avec la même
constance pour tous les types de protéines et de protocoles, suivant
la composition aminoacide et les additifs utilisés. Il est important
de noter que la fluorimétrie n’est pas une lecture au point final,
mais peut aussi être utilisée pour comparer des échantillons.
La chromatographie d’exclusion stérique ne tolère pas
les grandes quantités d’agrégats devant être injectées
sur la colonne. Certains des additifs utilisés sont agressifs et peuvent
endommager les phases stationnaires des colonnes. Le fait de colmater la colonne
entraîne une augmentation de la pression, des problèmes de reproductibilité
et une grande diminution de la durée de vie de la colonne.
La solution
Dans ces conditions, quelle est donc la solution ?
Novartis a utilisé un système de fractionnement par couplage
flux-force Eclipse™ et un détecteur de diffusion de la lumière
multiangles miniDAWN (tous deux conçus par Wyatt Technology, Santa
Barbara), associés à un Agilent HP1100 afin de fournir des capacités
de flux, dégazage, détection UV et d’échantillonnage
automatique.
Le dispositif Eclipse fonctionne sur la base du fractionnement par couplage
flux-force, mais la mise en œuvre technique rend le système aussi
simple à utiliser qu’une SEC ou une HPLC.
Fractionnement flux-force
La caractérisation macromoléculaire via FFF est un mécanisme
de séparation non destructeur en une seule phase servantà déterminer
les masses moléculaires et macromoléculaires et les particules
dans une solution. Comme pour la chromatographie classique, les composants
de l’échantillon subissent une élution à des temps
de rétention donnés qui sont liés à une série
de propriétés physiochimiques des espèces retenues. Cependant,
contrairement à la chromatographie classique, une séparation
FFF agit uniquement sur les propriétés hydrodynamiques et n’implique
aucune interaction avec une phase stationnaire.
La séparation est obtenue par le flux différentiel d’un
solvant dans un canal composé de deux plaques séparées
par une feuille séparatrice (« spacer ») ; les plaques
sont vissées ensemble. La plaque supérieure du canal est imperméable,
tandis que la plaque inférieure est perméable et composée
d’un fritté poreux. Une membrane d’ultrafiltration avec
une barrière de taille typique de 10kD recouvre le frittée afin
d’empêcher l’échantillon de sortir du canal.
Lors d’une première étape, l’échantillon
est introduit dans le courant du solvant qui est injecté dans le canal.
Dans le cadre d’une partie indispensable du processus de séparation,
l’échantillon est d’abord concentré dans la région
du canal proche du port d’injection. On y parvient en séparant
le flux du canal en deux composants, chacun d’entre eux entrant dans
le canal par les extrémités opposées. Les flux sont ajustés
de sorte qu’ils se rencontrent à proximité du port d’injection,
et, à ce stade, la direction du flux est perpendiculaire au fond poreux
du canal. L’échantillon est dirigé vers la paroi du fond
et concentré près de la membrane. La diffusion associée
au mouvement brownien crée un mouvement contraire. En quelques minutes,
un équilibre stationnaire est établi, dans lequel les deux forces
contrebalancent chaque taille de particule à une distance différente
de la membrane. Les particules de plus petite taille, présentant des
taux de diffusion supérieurs, et qui sont soumises à des forces
de friction inférieures, atteignent une position d’équilibre
plus loin de la paroi du fond. Les particules ou les molécules plus
grandes seront amenées à rester plus près de la paroi
du fond.
Lors de la deuxième étape impliquant l’élution,
le schéma du flux est rétabli pour s’écouler de
l’entrée du canal à la sortie et pour transporter l’échantillon
tout le long du canal. Le flux est laminaire, c’est-à-dire qu’il
présente un profil de vélocité parabolique et une vitesse
de transport supérieure à partir de la paroi du fond. Il s’agit
du même phénomène que dans une rivière où
les bateaux flottent vers l’aval plus rapidement au milieu du courant.
Les composants de l’échantillon sont comme des bateaux dans une
course ; les petites particules, plus éloignées de la «
plage », circuleront plus rapidement et subiront l’élution
en premier, avant les plus grandes. Il s’agit du phénomène
exactement inverse à celui de la séparation par exclusion stérique
ou perméation de gel (SEC/GPC) lors desquelles les grandes molécules
subissent l’élution en premier.
Le processus est rapide et doux du fait des faibles forces de cisaillement
et de l’absence de phase stationnaire. Par rapport à la SEC,
la FFF affiche une plus grande sélectivité et une plage d’application
aussi large.
Diffusion de la lumière multi-angles
(MALS)
Les détecteurs MALS utilisés dans cette étude sont les
seuls détecteurs absolus de diffusion de la lumière multi-angles
disponibles sur le marché. Cela signifie qu’ils mesurent la masse
molaire directement à partir de l’intensité de la lumière
diffusée, sans référence à une masse molaire standard
ou « connue ».
Dans l’application de détermination de la masse molaire d’un
monomère protéique et de détection des agrégats,
la sensibilité du détecteur de diffusion de la lumière
est capitale. Les protéines sont présentes en très faibles
concentrations et seuls quelques microlitres peuvent être utilisés
pour la mesure. Comme l’intensité de diffusion est proportionnelle
à la masse molaire des temps de concentration, le pic de diffusion
de la lumière résultant sera d’un ordre de magnitude inférieur
pour les monomères par rapport aux agrégats, car ces derniers
ont de fortes masses molaires de plusieurs millions de Daltons par rapport
à quelques dizaines de milliers de Daltons pour les protéines
natives.
Par conséquent, la tâche de détection est conséquente
et nécessite un photomètre de diffusion de la lumière
à la forte gamme dynamique. La technologie FFF s’est avérée
à la hauteur de ce défi et a fourni des données précises
pouvant être utilisées afin d’identifier les monomères
protéiques et les agrégats en un seul cycle de séparation.
La structure cellulaire unique associée à un banc optique et
une tête de lecture sophistiqués se sont avérés
capitaux dans le traitement d’aussi petits pics de diffusion de la lumière.
FFF-MALS
La technique FFF-MALS, alliant une séparation haute résolution
à une détection absolue des masses molaires, s’est avérée
plus qu’adaptée pour identifier et quantifier des protéines
monomériques repliées. Les cycles ont typiquement été
terminés en 12 minutes, produisant des résultats hautement reproductibles.
Les monomères et les agrégats ont pu être séparés
de manière fiable. Le système a été automatisé
en utilisant un échantillonneur automatique HPLC, et s’est avéré
assez sensible pour fournir de bons signaux avec des quantités aussi
infimes que 1 à 2 µg de protéine.
En utilisant la MALS, Novartis a été en mesure d’obtenir
des calculs de masse précis de monomères protéiques repliés–
typiquement, dans les 10 % de la valeur nominale. La quantification de rendement
a permis un classement des composant de la matrice de repliement afin d’évaluer
le protocole de repliement le plus productif pour une protéine spécifique.
Par exemple,
les monomères et les dimères peuvent être détectés
et quantifiés de la manière présentée dans la
figure ci-joint.
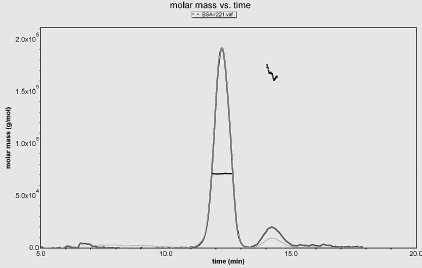
Résultats
Afin d’évaluer le précision, Novartis a réalisé
plusieurs cycles consécutifs d’une protéine et a vérifié
que la quantification du monomère/dimère était reproductible.
Dans une étude modèle, la méthode FFF-MALS a été
comparée à la quantification RPHPLC d’un monomère
replié. On a montré que tandis que la RP-HPLC rapportait des
valeurs protéiniques constantes, la méthode FFF-MALS était
en mesure de discriminer différents protocoles de repliement et montrait
des variations valides de la récupération du monomère.
Conclusion
La FFF basée sur le système Eclipse associéeà
la diffusion de la lumière multi-angles s’est avérée
une méthode solide et fiable de classement des conditions de repliement.
Bien que Novartis a testé plusieurs méthodes afin de déterminer
si les protéines s’étaient ou non repliée, la méthode
FFF a été de loin l’outil de caractérisation le
plus fiable et informatif. En outre, on a constaté que la FFF était
une excellente méthode de caractérisation des protéines
produites pour élucidation structurelle.
En utilisant la FFF-MALS, Novartis a replié plusieurs nouvelles protéines,
et a aussi significativement améliorée plusieurs de ses protocoles
de repliement. De plus, les résultats ont notablement varié
d’une protéine à l’autre, ce qui signifie que ce
n’est qu’en passant en revue la matrice de test que l’on
a pu trouver le protocole optimal. D’autres développements dans
l’écran de pliement semblent prometteur, et Novartis procéderaà
des modifications/ optimisation continuelles de l’écran. Celui-ci
devra aussi subir quelques expansions.
Nos remerciements à René Hemmig et Paul Ramage de Novartis Pharma
AG qui nous ont fournis les matériaux pour cet article.